Fragment building#
In this tutorial we will create a new dye-linker-nucleotide fragment which can be referenced by the FluorLabel PyMOL plugin. Before starting, make sure you have the following components installed on your machine:
FRETlabel
PyMOL 2.3 (PyMOL 2.4/2.5 crash upon
cmd.pair_fit)Antechamber (part of AmberTools19)
Refer to the installation page for specific instructions.
The PyMOL plugin FluorLabel fuses a fragment that consists of a modified nucleobase and a fluorophore connected by a flexible linker.
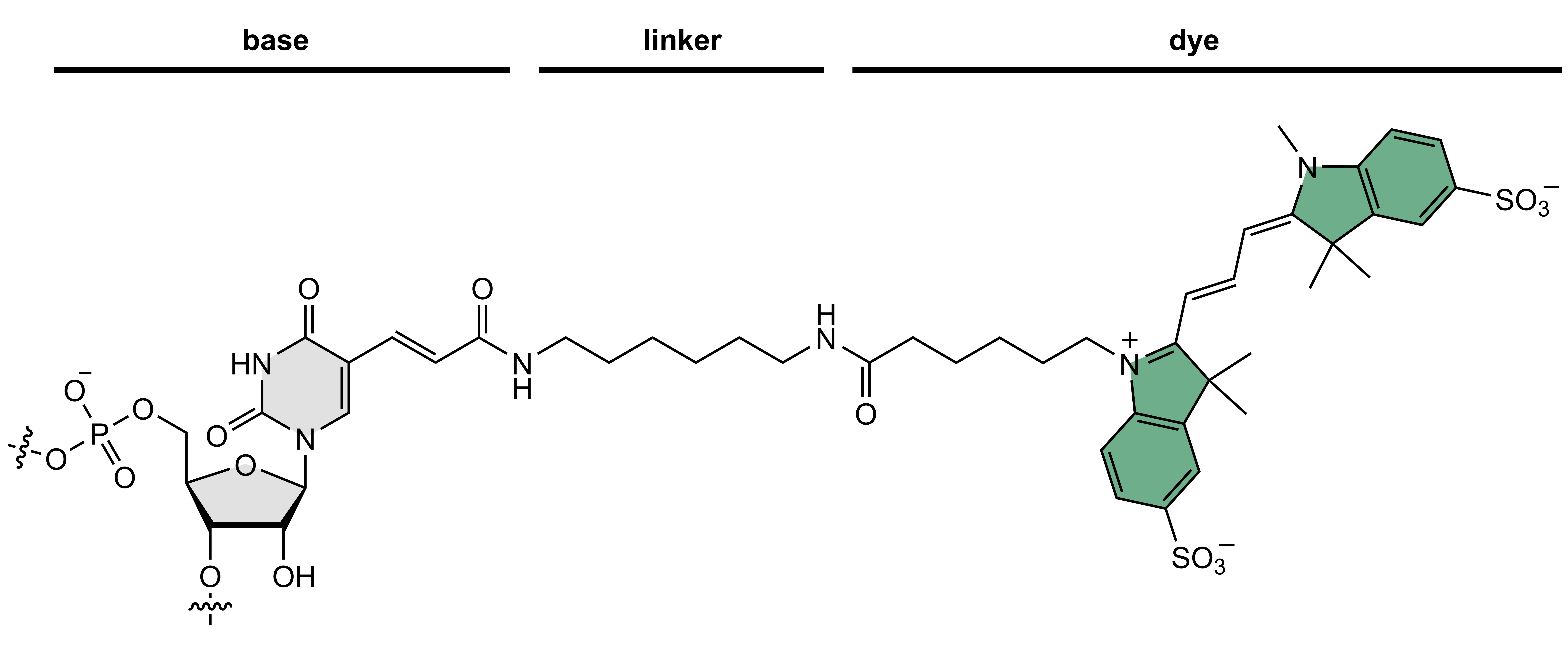
Pre-generated parameters in FRETlabel
Geometries, partial charges and force field parameters of selected dyes, linkers and nucleobases are distributed with FRETlabel. These files are located in the fragments/ and forcefields/ directories and have been generated using the code available here. To learn more about the specific attachment chemistries, have a look here.
The purpose of this tutorial is to demonstrate the process of generating a new dye-linker-base fragment. We will use a sulfonated Cy3 dye attached to the C5 of deoxythymine (see above) as an example.
The job mainly consists of two subtasks:
generating the geometries for the dye-linker-base fragment (i.e. a PDB file that can be referenced by FRETlabel)
deriving force field parameters for the new linker and attachment atoms/bonds/dihedrals
AMBER-DYES
A number of commonly used fluorophores has been previously parameterized in the AMBER force field for Gromacs by the Grubmüller lab (Graen et al. JCTC, 2014). The parameters are distributed through the AMBER-DYES package and are also included in FRETlabel. Dye parameters for the CHARMM package have also recently been released (Shaw, JCTC, 2020). Here we focus on deriving parameters for the AMBER force field. Note, however that once a dye-linker fragment (step 1) with appropriate force-field parameters (step 2) is generated it can be used by FRETlabel independent of the type of force field.
1. Building and paramerizing the linker#
The parameterization involves calculating partial charges as well as bond, angle and dihedral parameters for all the atomtypes. The parameters of the bases and the dyes are already available as part of the AMBER and AMBER-DYES force fields. However, those of the linker need to be generated from scratch or in analogy to existing parameters.
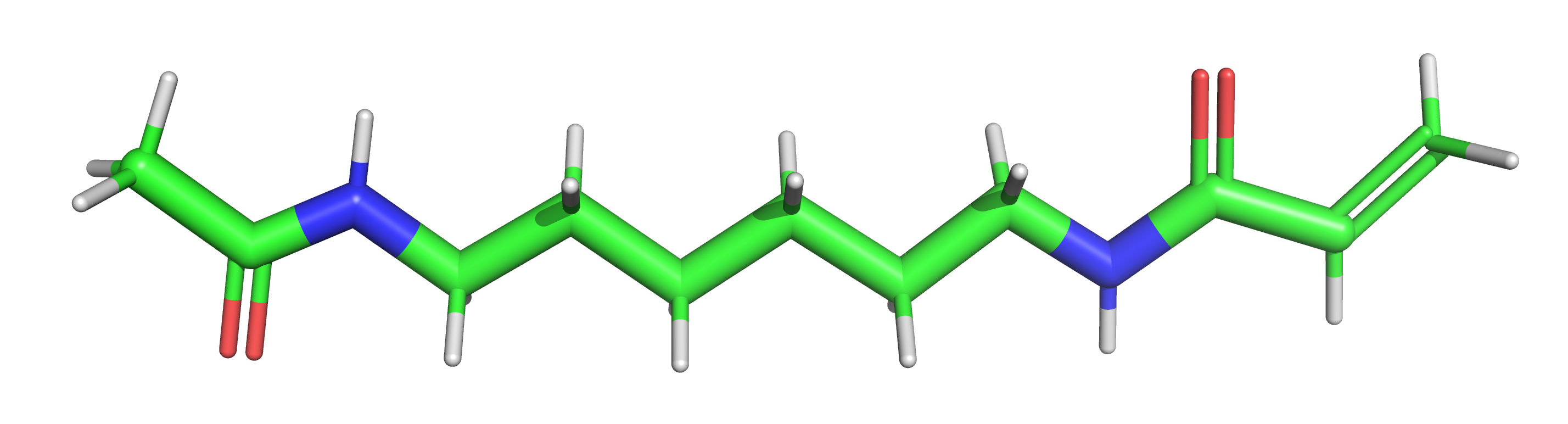
We will start by calculating partial charges of the linker by doing a RESP (Restrained ElectroStatic Potential) fit. For this labeling chemistry we use the following 6-carbon linker capped at each both ends with either a methylene or an acetylene (ACE) group to fill the bond valencies and to make the net charge of the spacer equal zero. The capping groups are chosen to best mimic the actual charge distribution when the linker is fused to the dye and the nucleotide.
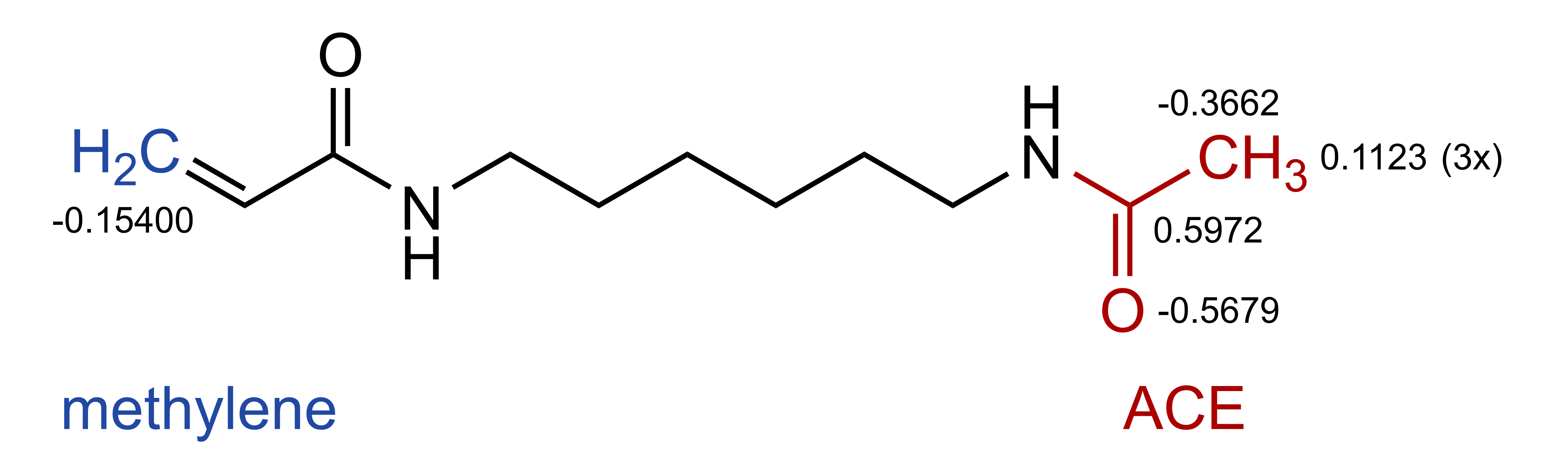
The partial charges of the ACE group have been derived when the AMBER-95 force field was designed (Cornell et al. JACS 1995) and are itself summing up to zero. The methylene charges will be determined as part of the charge fitting procedure with a group charge constraint of -0.15400. This group constraint equals the charge difference between a normal deoxythymine and one where two of the three methyl hydrogens on C5 are removed (partial charge of H71 and H72 is 0.77200 each).
1.1 Build the capped linker with PyMOL#
First, we create the linker with correct bond valencies using the Builder tool of PyMOL. Fragments can be added alltogether by Build→Fragment→Acetylene.
Atom naming
Rename the atoms in a logical way such that there is no overlap with the atom names of the nucleotide (atoms of the linker and the dye can have the same names because there residue name is different).
Tip
Mol2 files have the advantage that bond valencies are encoded in the format. You can emulate this behavior with PDB files by creating double CONECT entries, however this feature is not recognized by many programs and often leads to error messages instead.
1.2 Geometry optimization and ESP calculation with Gaussian#
Next, we will do a geometry optimization and calculate an electrostatic potential (ESP) using GAUSSIAN (or alternatively GAMESS). A Gaussian input file can be created via Antechamber:
name=MLE_capped
input_folder='in/'
output_folder='out/'
cd fragments/2_linkers/MLE/
antechamber -i "$input_folder/$name".mol2 -fi mol2 -o "$output_folder/$name".gin -fo gcrt -gv 1 -ge "$output_folder/$name".gesp -ch "$output_folder/$name"_opt -nc 0
-iinput filename-fiinput file format-ooutput filename-fooutput file format (gcrt = Gaussian Cartesian)-gvadd keyword to generate gesp file (1 = yes, for Gaussian09)-gegaussian esp filename generated by iop(6/50=1), default is g09.gesp-chcheck filename for Gaussian, default is ‘molecule’-ncnet charge
To speed up the calculation we will perform the geometry optimization at the B3LYP/6-31G* level of theory followed by the ESP calculation using Hartree Fock (HF/6-31G*). For this purpose, we will slightly modify the Gaussian input file and allow it to run on multiple cores (with the nproc keyword).
nproc=12
sed 's/#HF.*/\#P b3lyp\/6-31G\* Opt/g' < "$output_folder/$name".gin | sed '/iop/d' | sed '/.*gesp/d' | sed "/--Link1--/ a %nproc=$nproc" > "$output_folder/$name"_b3lyp_opt.gin
sed '/^[[:space:]]*[A-Z]/d' < "$output_folder/$name".gin | sed 's/SCF/Geom=check SCF/g'| sed 's/\(\%chk=.*\)opt/\1esp/g' | sed "/--Link1--/ a %nproc=$nproc" > "$output_folder/$name"_hf_esp.gin
Run the geometry optimization:
g09 < "$output_folder/$name"_b3lyp_opt.gin > "$output_folder/$name"_b3lyp_opt.gout && cp "$output_folder/$name"_opt.chk "$output_folder/$name"_esp.chk
Note
Make sure that charge and multiplicity are compatible (check log files in case of segmentation errors).
Calculate the electrostatic surface potential using the checkpoint file of the geometry optimization as start coordinates.
g09 < "$output_folder/$name"_hf_esp.gin > "$output_folder/$name"_hf_esp.gout
Note
Remove the coordinates from the original file, in order to use the checkpoint file from the geometry optimization.
1.3 Partial charge fitting with RESP#
Use Antechamber to convert the mol2 file of the linker into an Antechamber file (.ac) which can be read by respgen.
antechamber -i "$input_folder/$name".mol2 -fi mol2 -o "$output_folder/$name".ac -fo ac -pf yes -nc 0
Create a DTM_capping_group.dat file to define which atoms should be assigned a fixed charge and which atoms to group together with a constraint. As mentioned above, we will fix the charges of the ACE atoms according to the AMBER force field and apply a group contraint on the methylene cap.
// ACE cap fixed to the charge of AMBER99ff (format: CHARGE <partial_charge> <atom_ID> <atom_name>)
CHARGE 0.5972 10 C16
CHARGE -0.3662 11 C17
CHARGE -0.5679 13 O98
CHARGE 0.1123 32 H97
CHARGE 0.1123 33 H96
CHARGE 0.1123 34 H95
// Group constraint for methylene cap (format: GROUP <number_atoms> <net_charge>)
GROUP 3 -0.15400
ATOM 1 C7
ATOM 16 H01
ATOM 17 H02
Run the RESP fitting (see below for information on the individual steps):
capping_group=MLE_capping_groups.dat
n_atom=`awk '$1 == "GROUP" {print $2}' "$input_folder/$capping_group"`
group_constraint=`awk '$1 == "GROUP" {print $3}' "$input_folder/$capping_group"`
respgen -i "$output_folder/$name".ac -o "$output_folder/$name".respin1 -f resp1 -a "$input_folder/$capping_group"
respgen -i "$output_folder/$name".ac -o "$output_folder/$name".respin2 -f resp2 -a "$input_folder/$capping_group"
#since respgen rounds the group constraint to three decimals replace it with the value from the capping group
sed -i "s/$n_atom.*\..*/$n_atom $group_constraint/g" "$output_folder/$name".respin1
sed -i "s/$n_atom.*\..*/$n_atom $group_constraint/g" "$output_folder/$name".respin2
espgen -i "$output_folder/$name"_hf_esp.gout -o "$output_folder/$name"_hf_esp.esp
mv QIN "$output_folder"/
resp -O -i "$output_folder/$name".respin1 -o "$output_folder/$name".respout1 -e "$output_folder/$name"_hf_esp.esp -q "$output_folder"/QIN -t "$output_folder"/qout_stage1 -p "$output_folder"/punch1 -s "$output_folder"/esout1
resp -O -i "$output_folder/$name".respin2 -o "$output_folder/$name".respout2 -e "$output_folder/$name"_hf_esp.esp -q "$output_folder"/qout_stage1 -t "$output_folder"/qout_stage2 -p "$output_folder"/punch2 -s "$output_folder"/esout2
antechamber -i "$output_folder/$name".ac -fi ac -o "$output_folder/$name"_resp.mol2 -fo mol2 -c rc -cf "$output_folder"/qout_stage2 -pf yes -at amber
In brief the above code block does the following:
Respgen generates the input files for two-stage RESP fitting (
-fspecifies the stage (resp1, resp2) and-areads in the frozen and group charges) along with aQINfile for the charge constraints.Espgen extracts the ESP from a Gaussian output file.
The RESP fit is performed in two stages: in the first all atoms are allowed to vary, in the second degenerate hydrogens are constrained to have an equal charge. Apart from the input (
-i) and output (-o), additional flags include:-Ooverwrite existing files-eESP file (input)-qcharge constraints (input, QIN)-tcurrent charges (output)-psynopsis of results (output)-sESP for new charges (output)
Finally, Antechamber combines the RESP charges into a new mol2 file (
-ccharge method rc = read in charge,-cfcharge filename,-pfremove intermediary files,-atatom type).
1.4 Fusing base, linker and fluorophore#
Now that we have all three components (the nucleobase, the linker and the dye all in mol2 format) we can fuse them together using PyMOL.
First, we fuse the base and the linker together. Use the 3-Button Editing mode of PyMOL to rotate the fragments with respect to each other to minimize sterical clashes.
Execute the following code from the PyMOL shell
import fretlabel as fl
names_methylene = ['C7','H01','H02']
base_resn = ('deoxythymidine', 'DTM')
cmd.reinitialize()
cmd.load('fragments/1_bases/out/deoxythymidine.mol2')
cmd.load('fragments/2_linkers/MLE/out/MLE_capped_resp.mol2')
cmd.remove('MLE_capped_resp and name {}'.format('+'.join(str(i) for i in names_methylene)))
cmd.remove('deoxythymidine and (name H71 or name H72)')
cmd.fuse('deoxythymidine and name C7', 'MLE_capped_resp and name C8 and resn MLE')
cmd.delete('deoxythymidine')
cmd.alter('all', 'type="ATOM"')
cmd.alter('all', 'elem=""') # PyMOL struggles with atom type definitions in mol2 files, therfore let PyMOL guess the elements itself
cmd.set_name('MLE_capped_resp', base_resn[1])
cmd.set_title(base_resn[1],1,base_resn[1])
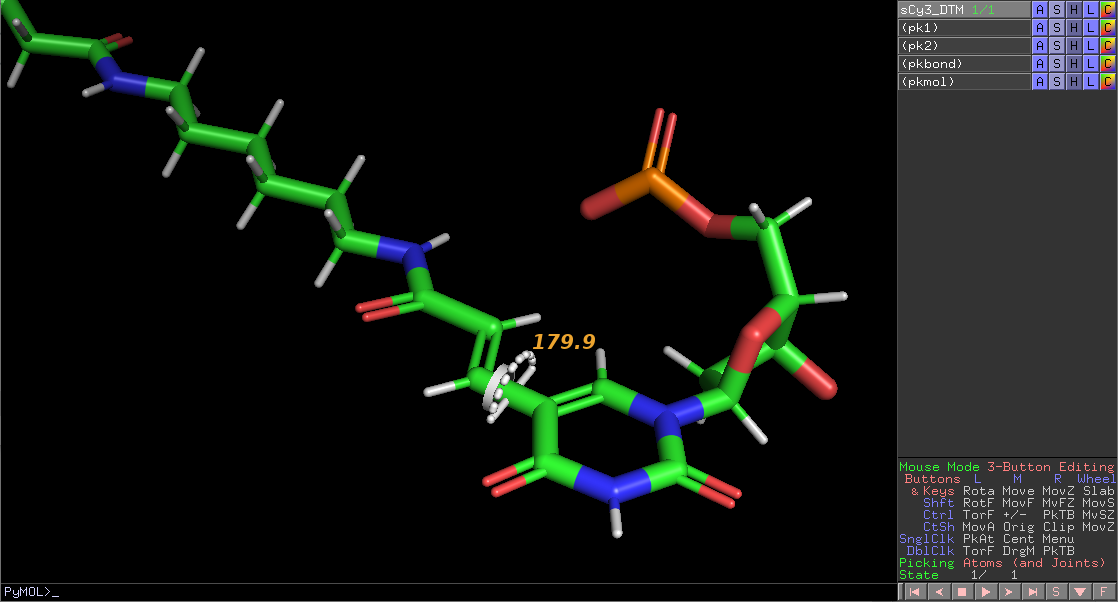
Update the valency of the bond between the linker and the base.
cmd.unbond('resn MLE and name C8', 'resn DT and name C7')
cmd.bond('resn MLE and name C8', 'resn DT and name C7', 2)
Save the base-linker as a mol2 file under fragments/4_base_linkers/
fl.ff.pymol_save_molecule(f'fragments/4_base_linkers/{base_resn[1]}.mol2', base_resn[1], 'mol2', overwrite=False)
Attach the dye to the base-linker and save the whole base-linker-dye fragment as a pdb file under src/fretlabel/dyes/ where it can be accessed by the PyMOL plugin FRETlabel.
dye = 'C3W'
fl.ff.pymol_couple_dye2baselinker(f'fragments/3_dyes/{dye}.mol2', f'fragments/4_base_linkers/{base_resn[1]}.mol2', 'C99', ['C17', 'O98', 'C16'],['O98', 'C16', 'C17', 'H95', 'H96', 'H97'])
fl.ff.pymol_save_molecule(f'src/fretlabel/dyes/{dye}_{base_resn[1]}.pdb', f'{dye}_{base_resn[1]}', 'pdb', overwrite=False)
Finally, add a new entry to src/fretlabel/dye_library.json and indicate the filename of your fragment, the dye, the base, the position and the alignment method (base or backbone).
[
{"filename":"C3W_DTM",
"dye":"sCy3",
"base":"DT",
"position":"internal",
"alignment":"base"}
]
2. Update the force field#
We will now create a GROMACS topology (-o) of our new base-linker fragment using the AnteChamber PYthon Parser interfacE (Acpype). This time we will choose AMBER as the atom type (-a) and read in the previously calculated RESP charges (-c). We first need to rename the base (DT) and the linker (MLE) to DTM with sed because Acpype only accepts one residue name per molecule.
cd fragments/5_acpype
filename=../4_base_linkers/DTM.mol2
base=DT
linker=MLE
base_linker=DTM
sed "s/${base}1/${base_linker}1/g" "$filename" | sed "s/${linker}1/${base_linker}1/g" > "${base_linker}"_ff.mol2
acpype -i "${base_linker}"_ff.mol2 -o gmx -n -1 -a amber -c user
Acpype generates an itp file as well as force field modification (frcmod) file.
The itp file lists all atoms, bonds, angles and dihedrals (both proper and improper) present in the molecule.
The frcmod file lists any atomtypes, bondtypes, angletypes and dihedraltypes which are present in the molecule but missing in the force field (here AMBER because of the
-aflag in the acpype call).
Note
The frcmod file is formatted for use with the AMBER MD programs LEaP and Sander. The file and units are converted internally by fl.ff.Parameter.read_frcmod() to the GROMACS format in the next step.
import fretlabel as fl
%cd ../../
baselinker_itp = fl.ff.Molecule.read_molecule('fragments/5_acpype/DTM_ff.acpype/DTM_ff_GMX.itp', 'FRETLABEL')
# change the atom type of O3' from O to OS since this residue is internal and not terminal
baselinker_itp.change_type('O3\'', 'OS')
for a in ['O98', 'C16', 'C17', 'H95', 'H96', 'H97']:
baselinker_itp.remove_atom(a)
baselinkers_ff = fl.ff.Parameters.read_frcmod('fragments/5_acpype/DTM_ff.acpype/DTM_ff_AC.frcmod', fretlabel_itp.atomtypes_molecule)
Next, we load in the force field parameters from AMBER-DYES
amberdyes_ff = fl.ff.Parameters.read_amberdyes(['forcefields/2_amberdyes/ffbonded_amberdyes.itp', 'forcefields/2_amberdyes/ffnonbonded_amberdyes.itp'])
The atoms involved in the bond, angles and dihedrals between linker and fluorophore need to be manually specified because the atom types from AMBER-DYES (for dye) and AMBER (for custom linker) are slightly different. The parameters are defined by analogy to those already present in AMBER-DYES.
atoms_amberdyes = {'bondtypes' : [['ng', 'cg']],
'angletypes': [['c3g', 'ng', 'cg'],
['hng', 'ng', 'cg'],
['ng', 'cg', 'og'],
['ng', 'cg', 'c3g']],
'propertypes' : [['c3g', 'c3g', 'cg', 'ng'],
['hcg', 'c3g', 'cg', 'ng'],
['c3g', 'cg', 'ng', 'hng'],
['og', 'cg', 'ng', 'hng'],
['c3g', 'cg', 'ng', 'c3g'],
['og', 'cg', 'ng', 'c3g']]}
atoms_linker = {'bondtypes': [['N', 'cg']],
'angletypes': [['CT', 'N', 'cg'],
['H', 'N', 'cg'],
['N', 'cg', 'og'],
['N', 'cg', 'c3g']],
'propertypes': [['c3g', 'c3g', 'cg', 'N'],
['hcg', 'c3g', 'cg', 'N'],
['c3g', 'cg', 'N', 'H'],
['og', 'cg', 'N', 'H'],
['c3g', 'cg', 'N', 'CT'],
['og', 'cg', 'N', 'CT']]}
specialbond_ff = fl.ff.Parameters.read_specialbond(amberdyes_ff, atoms_amberdyes, atoms_linker)
Append the parameters of the linker (in baselinkers_ff) and the bond linking the dye and the linker (in specialbond_ff) to the dye parameters (in amberdyes_ff)
amberdyes_ff.append(baselinkers_ff)
amberdyes_ff.append(specialbond_ff)
Write new ffnonbonded.itp and ffbonded.itp files of the combined forcefield (amber14sb, amberdyes_ff, baselinkers_ff and specialbond_ff) into a directory 4_fretlabel/.
amberdyes_ff.add2ff('forcefields/3_amber14sb_DTM', 'forcefields/4_fretlabel/')
Write a residue topology file (rtp) for the linker atoms, bonds and proper dihedrals
fl.ff.write_rtp('forcefields/4_fretlabel/fretlabel_DTM.rtp', [baselinker_itp])
Update the specbond.dat file with the bond between the dye and the linker.
fl.ff.update_specbond('C3W C99 1 DTM N99 1 0.160 C3W DTM', 'forcefields/2_amberdyes/specbond_amberdyes.dat', 'forcefields/4_fretlabel/specbond_DTM.dat')
Update the residuetypes.dat file with the types of the the dye and the new fragment.
fl.ff.update_residuetypes('C3W DNA', 'forcefields/2_amberdyes/residuetypes_amberdyes.dat', 'forcefields/4_fretlabel/residuetypes_DTM.dat', overwrite=True)
fl.ff.update_residuetypes('DTM DNA', 'forcefields/4_fretlabel/residuetypes_DTM.dat', 'forcefields/4_fretlabel/residuetypes_DTM.dat', overwrite=True)