Calculating ACVs#
Accessible contact volumes and FRET calculations can be done directly from PyMOL. The PyMOL plugin allows the user to visually interact with the molecular viewer while exploring optimal sites for labeling a biomolecule of interest. This tutorial will guide you step-by-step through the workflow.
Start by opening the FRETraj GUI from the Plugin Menu.
Note
Upon first startup, FRETraj asks the user to define a root directory where all output files (ACVs, label and FRET parameters) will be stored (e.g. C:/Users/fsteffen/DNA-hairpin/fretraj/)
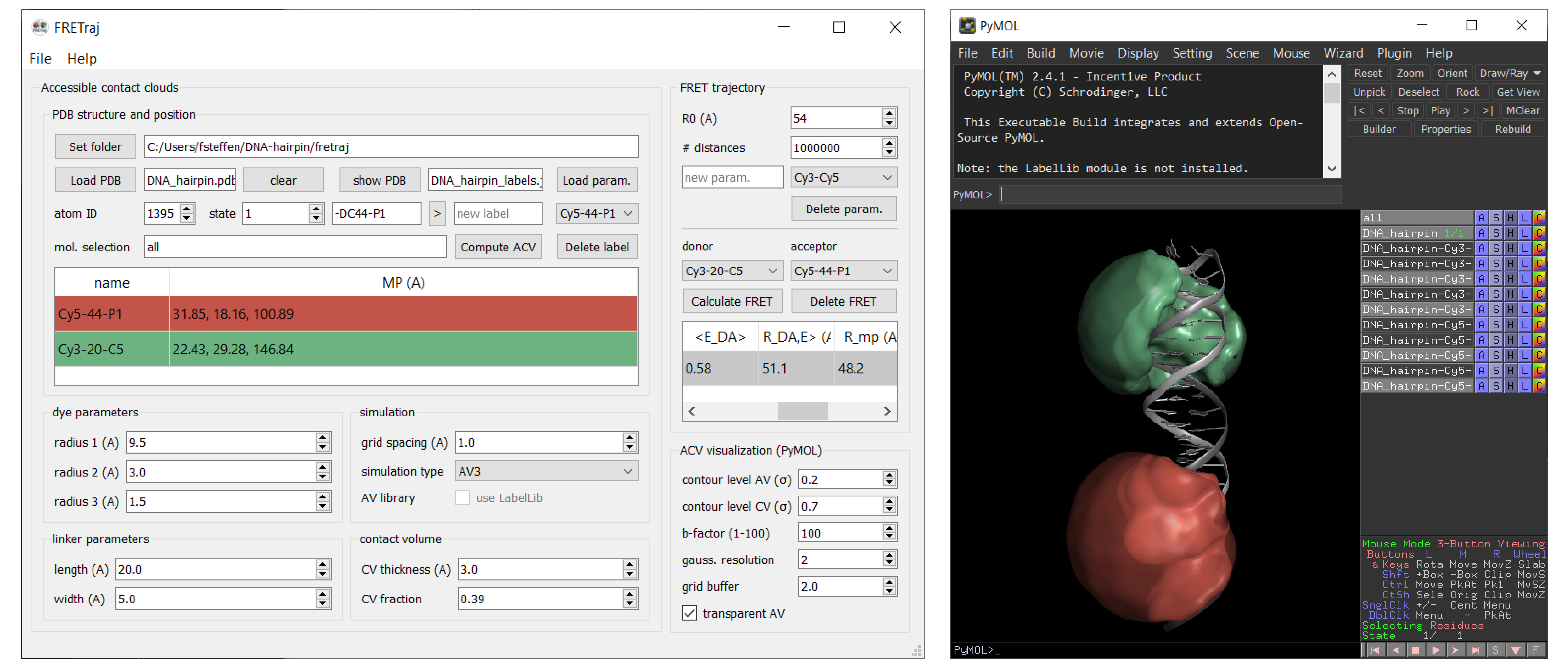
Fig. 3 PyMOL with FRETraj’s graphical user interface (left) and molecular viewer (right).#
Tip
Within FRETraj you can this an other demo projects by going to Help → Load Example.
The graphical user interface (GUI) is divided into two sections (1) Accessible contact clouds and (2) FRET trajectory. The labeling positions are defined in the panel PDB structure and position . The panels below specify the dye, linker, simulation and contact volume parameters. All these settings are saved into the parameter file.
In the following the most important settings and functionalities of FRETraj for simulating accessible-contact volumes and predicting FRET efficiencies are described.
Load PDB and define dye positions#
Set folder: define the root folder where the ACVs and the parameter file are saved
Load PDB: import a single or multi-state PDB/CIF file
Show Text: Show the PDB text file in a integrated viewer to help with the atom ID selection
Hint
If you already have a JSON-formatted parameter file you can load this file with the Load Param button and proceed directly to the last step compute ACV.
atom ID: select the attachment atom on the biomolecule by its serial atom ID (i.e. the ID of the atom given in the DPB file in columns 7-11, see Note below).
Note
PyMOL uses the following identifiers:
idrefers to the atom identifier (number present in columns 7-11 of the PDB file) which correspponds to mdtraj’sserial(see below). Usually PDBs start withid=1but this may vary if the PDB is sliced.indexrefers to the 1-based index)rankrefers to the 0-based index which corresponds to mdtraj’sindex(see below)
mdtraj exposes two atom selection options:
serialrefers to the atom identifier (number present in columns 7-11 of the PDB file), e.g.[a.serial for a in dna.topology.atoms][0]returns1.indexrefers to the 0-based indexe.g.[a.index for a in dna.topology.atoms]returns0. Note that, when usingatom_slicethe selection is re-indexed, i.e.[a.index for a in dna.atom_slice([2]).topology.atoms]returns0but theserialis maintained, i.e.[a.serial for a in dna.atom_slice([2]).topology.atoms]still returns3, given that the original PDB started withid=1).
Internal indexing in FRETraj
In FRETraj we use
attach_idto refer to the PyMOL’s atomid/ mdtraj’sserial. Internally, we match the atom serialidto mdtraj’s 0-basedindex(attach_id_mdtraj). This ensures consistency inattach_idwhen the PDB is sliced.state: select a different state of the PDB file
Add the new position to the label dropdown by pressing on ▶️
Note
If you wish to remove a label position from the list press on Delete label (this does not remove any associated ACV PDB files from the root folder)
mol. selection: consider only a subregion of your biomolecule while computing ACVs. While this may speed up the calculation, usually you want to leave this at all (default) for simplicity.
Define dye and linker dimensions#
Dye parameters: Set the radii of the dye (see section Accessible volume)
Linker parameters: Set the length and width of the carbon linker
Simulation:
grid spacing: distance between two grid points
simulation type: select between a single-radius (AV1) or a three-radii (AV3) dye probe
AV library: if LabelLib has been installed, you may select use LabelLib to speed up the calculations. Otherwise a pure Python version of the ACV algorithm is used.
Contact volume: Choose the thickness and relative fraction of the contact volume
Note
The parameter file is dynamically updated whenever a new label is added/removed or a setting is changed.
Compute ACV: Start the calculation. The ACV is automatically saved to the selected root folder. Once the calculation has finished the ACV cloud is displayed in the molecular viewer and the mean position (MP) is indicated in the table